
미국 식품의약국(FDA)은 미국에서 유통되는 거의 모든 식품, 의약품, 의료기기, 화장품 등의 안전성을 관리한다. 예컨대 농산물은 미국 농무부(USDA)가 관리하지만 슈퍼마켓에서 판매하는 가공식품(processed food, USDA와 공동관리) 및 식이보충제(Dietary Supplement, 건강보조식품, 건강기능식품과 비슷)은 전적으로 FDA가 관리한다.
의약품(전문의약품 및 일반의약품) 및 의료기기(병원용 및 가정용) 일체와 화장품(샴푸, 치약 등도 포함)도 FDA가 관리한다.
FDA는 제품이 미국 시장에 출시되기 전에 반드시 일정한 절차를 거치도록 요구한다. 의료기기의 경우 등록(Registered), 승인(Approved), 허가(Cleared)라는 판매 승인 절차가 제품의 수준(안전성, 위험도)에 따라 나뉘어져 있어 이에 대한 명확한 이해가 필요하다.
특히 국내기업이 미국에서 승인받은 대부분의 인증은 거의 대부분 ‘등록’ 또는 ‘허가’에 불과한데 이를 마치 정식 ‘승인’을 받은 것처럼 호도하는 경우가 있어 이를 분간해야 한다.
FDA는 의료기기의 위험도에 따라 1등급, 2등급, 3등급으로 분류한다. 1등급은 메스, 거즈, 마스크 등 건강과 안전에 심각한 위협을 주지 않는 단순한 기능을 가진 최소한의 위험을 내포한 의료기기다. 2등급은 휠체어, 콘택트렌즈, 콘돔, 정형외과 고정심지(스테이플) 같은 경미험 위험이 있는 제품으로 인체의 건강과 안전에 직접적인 영향을 끼치는 의료기기다. 3등급은 심박 조율기, 인공 판막, 로봇수술기 등 인체에 이식되거나 생명 유지에 사용되는 매우 큰 위험성을 갖는 의료기기를 말한다.
FDA 등록은 FDA에 해당 의료기기를 만든 기업의 정보, 제품 정보(재원), 공장 정보를 올리는 것이다. 모든 의료기기가 등록 대상이다. FDA 등록 없이 미국에 의료기기를 생산, 수입, 유통하는 것은 불법이다.
미국에서 ‘활동’ 또는 상업적 유통을 시작한 후 30일 이내에 FDA에 시설을 등록해야 한다. 외국 시설은 미국으로 제품을 수출하기 전에 등록해야 하며, 미국 내 시설은 수입하기 전에 등록해야 한다.
FDA 등록은 해마다 경신돼야 한다. 10월 1일에서 12월 31일 사이에 기 등록한 정보를 업데이트해야 한다. 특별한 하자가 없는 한 등록은 수리가 된다. 이는 기본적인 것으로 정식 승인과는 직접적인 관련이 없다. 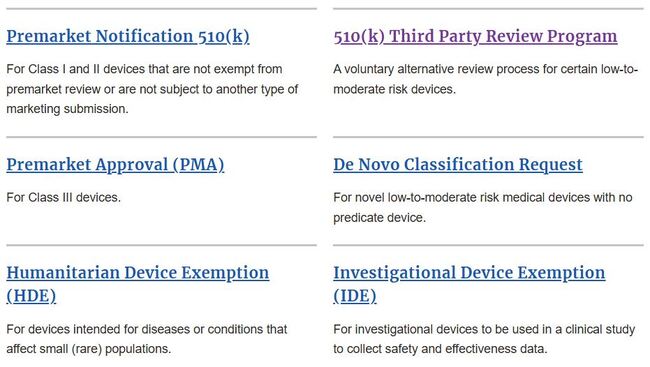 미국에서 의료기기 시판전 사용을 위해 규정한 6가지 형태의 시판전 심사 경로(출처 FDA 홈페이지)
미국에서 의료기기 시판전 사용을 위해 규정한 6가지 형태의 시판전 심사 경로(출처 FDA 홈페이지)
FDA 허가(Cleared)는 정의 이전에 허가된 의료기기, 즉 ‘기재 내용’과 실질적으로 동등함을 입증하는 2등급 의료기기에 부여한다. FDA는 1976년에 개정된 의료기기법에 따라 기존 기기 목록을 기반으로 ‘허가’ 카테고리를 분류했다. 오늘날 미국에서 판매되는 모든 2등급 제품은 1976년에 개정된 분류 기준을 기반으로 허가가 이뤄진다.
2등급 기기는 승인(Approved) 대신 510(k)라는 ‘시판 전 신고’(Pre-market notification)절차를 거쳐 FDA ‘허가’를 받는다. 이는 이미 합법적으로 판매 중인 기존 기기와 유사하기 때문에 ‘안전성도 실질적으로 동등하다’는 서류 제출을 통해 통과할 수 있다.
FDA에서 규제하는 ‘시판 전 심사제도’는 크게 네 가지로 구분한다. △510(k) - 시판 전 신고(Premarket Notification) △HDE(Humanitarian Device Exemption) - 인도적 예외 조항 적용 의료기기 △드노보(De Novo) - 신기술 의료기기 △PMA(Premarket Approval) - 시판 전 승인 등이다.
510(k)는 유통하려는 의료기기가 사용 목적, 기술적 특성, 성능 감사 등의 항목에서 기존의 장비와 실질적으로 동등하므로 시판해도 안전하다는 것을 증명하는 절차다.
미국 식품, 의약품 및 화장품법(Food, Drug and Cosmetic Act, FD&C법)의 510(k) 부분에 따르면 의료기기 업체는 최소 90일 전에 의료기기를 판매하려는 의도를 FDA에 제출해 허가를 받아야 한다. 이를 시판 전 신고(Premarket Notification, PMN) 또는 510(k)라고 한다. FDA는 신규 의료기기가 기존 의료기기와 동일한지를 판단해 ‘허가’를 한다. 일반적으로 검토 기간으로 5개월이 소요되며, 제출 건수의 67%(의약품 포함)가 자료 보완 요구를 받는다고 한다. 수정을 빠르게 끝내고 싶으면 급행 수수료를 내고 ‘Special 510(k) route’ 절차를 밟으면 30일 이내에 510(k) 절차를 완료할 수 있다.
이전에 분류되지 않은 ‘새로운’(De Novo) 의료기기(1976년 5월 28일 이전에 상업적으로 등록 판매되지 않음)는 따로 구분해 심사한다. De Novo 분류 신청(De Novo Classification Request)은 일반규제만으로 또는 일반규제 및 특별규제만으로 의도된 용도에 대한 안전성과 유효성을 합리적으로 보장할 수 있지만, 법적으로 시판되는 선행 의료기기가 없는 신규 의료기기를 따로 분류해 별도의 시판절차를 부여하는 것을 말한다. 보통 1등급 또는 2등급으로 분류된 의료기기에 대해 시판 전 통지(510(k) 제출)을 위한 사전 준비로 이 절차를 활용한다.
FDA가 옵션으로 둔 드노보 요청을 제출할 수 있는 경우는 두 가지다. △ 510(k) 검토를 받고 FDA가 해당 기기가 선행 기기와 ‘실질적으로 동등하지 않다’(Not Substantially Equivalent)고 결정한 경우 △510(k) 제출 대신 사용 가능한 선행 기기가 없는 것이 확실한 경우다.
드노보 요청에서 다음 요건을 충족해야 드노보 경로를 인정받을 수 있다. △해당 기기가 FD&C법 제513(a)(1)항에 따라 1등급 또는 2등급으로 분류되는 기준을 충족한다는 것을 증명한다. 즉, 일반규제 또는 일반 및 특수 규제가 해당 기기의 안전성과 효능에 대한 합리적인 보장을 제공한다는 것을 증명한다. △의료기기의 건강에 대한 모든 잠재적 위험과 잠재적 이익을 알리고, 모든 잠재적 위험을 완화하는 데 필요한 조치를 설명하며, 일반규제 또는 일반 및 특별 규제를 적용해 기기의 안전성과 효능을 어떻게 보장할 수 있는지 설명한다.
FDA는 드노보 제출 이후 120일 이내에, 해당 기기가 1등급인지 2등급인지를 판단해 새로운 제품코드와 규정 번호를 발부한다. 즉 드노보 경로로 허가를 받는다. 드노보 경로가 거부된다면 3등급으로 분류돼 엄격한 PMA를 거쳐 정식 승인을 얻어야 한다.
인도적 예외조항(HDE) 의료기기는 인도적 차원에서 해당 의료기기의 유효성을 입증하는 자료를 따로 제출할 필요가 없는 것을 말한다. 1990년에 설정된 이 조항은 희귀질환에 대한 의료기기에 주로 적용된다. 유효성을 과학적으로 입증할 필요는 없으나 예상되는 이점에 대해 기술해야 한다.
이밖에 연구자 주도 임상을 위한 사전 사용 허용(Investigational Device Exemption, IDE)가 있다. 의료기기의 안전성과 유효성을 확인하기 위한 연구 차원의 임상시험을 위해 사전에 FDA 허가를 받는 것으로, 공식 허가나 승인과는 사실상 무관하다.
가장 고강도 사전심사가 3등급 의료기기에 대한 PMA 절차다. PMA는 엄격하고 포괄적인 안전성 및 유효성 테스트(인체 임상시험)를 의무화하고 있다. 공식 승인 전 의무적으로 3등급 의료기기와 안전성와 유효성을 보장하기 위한 PMA 절차를 거쳐야 한다.
FDA 정식 승인(Approved)의 의미
3등급 의료기기는 사용자에게 높은 위험도를 수반하므로 FDA는 가장 면밀한 감독을 해야 한다. FDA는 시판 전 승인(PMA) 절차를 가챠 3등급 의료기기를 정식으로 ‘승인’하게 된다. PMA 신청의 목적은 해당 의료기기가 사용자에게 안전하며 의도된 기능을 수행하는지 증명하는 것이다.
FDA 승인은 PMA 신청 절차로만 받을 수 있다. 이 절차를 거치지 않은 의료기기가 ‘FDA 승인’이라고 언급하거나 판매해서는 안 된다.
일부 2등급 기기도 PMA 신청 절차를 거쳐야 한다. 승인된 기기와 실질적으로 동등성을 입증한 2등급 기기는 510(k)를 통해 FDA 허가를 받을 수 있지만, 기존 승인된 기기와 동등성을 입증하지 못한 기기는 PMA 절차를 따르거나, 자격이 되는 경우 드노보 요청을 제출한다.
결론적으로 등록은 의료기기의 미국내 유통을 위한 신고사항이자 의무사항이다. 허가는 의료기기를 등록한 뒤 FDA 검토하고 510(k) 절차를 밟은 것을 의미한다. 다시 말해 주로 1등급 또는 2등급 의료기기로서 기존 의료기기와 별 차이가 없는 경우에 해당한다.
승인은 3등급 의료기기가 합법적으로 판매되는 과정으로 엄격한 검토 및 승인 절차를 거쳐야 한다. 시판 전 승인(PMA) 또는 예외 조항 적용 의료기기(HDE)를 제출하면 FDA가 검토해 해당 기기를 ‘승인’ 한다.
참고로 모든 신규 전문약(처방약, 신물질은 물론 제형개량, 신규 복합성분 조합)은 허가가 아닌 승인을 받아야 한다. 다만 제네릭은 약식의 신약승인신청(abbreviated new drug application, ANDA)를 제출하는데 통과할 경우 이를 승인(Approval) 또는 공인(authorization)이라고 한다. 미국에서는 제네릭이라 할지라도 신약에 못잖은 깐깐한 서류심사를 거쳐야 승인이 나온다.
드노보 경로 의료기기는 미국에서 합법적으로 판매되기 전에 FDA의 승인을 받아야 한다. 신규성을 가진 의료기기로서 1등급 또는 2등급의 위험성이 인정된다고 간주할 경우에 시판을 인정해주는 게 드노보 경로로, 법적으로 허가가 아닌 승인(approval)으로 간주한다.
따라서 독자가 기사를 읽을 때 510(k) 절차를 완료한 것을 ‘승인’이라고 받아들이면 잘못된 것이다. 이는 시판 전 신고로 FDA ‘승인’이 아니라 ‘허가’를 통지받은 것을 의미한다. 510(k) 허가를 PMA 통과(정식 승인을 위한 직전의 절차)로 오인하게끔 언급하는 건 위법사항에 해당할 수 있다.
예컨대 애플와치는 FDA에서 2등급 의료기기로 ‘510(k) 허가’를 받았는데 언론이나 독자는 FDA ‘승인’으로 오해하는 경우가 많다.
미국 시장의 의료기기 대부분은 510(k) 절차를 통해 허가받고 있으며, 임상시험이 필요 없고 감독도 필요로 하지 않는다. 환자단체와 정부 감시기관은 510(k) 시스템을 매우 비판적으로 평가했다. 위험한 기기들이 510(k)로 허가받고 시장에 나와 이를 의심하지 않고 사용한 환자를 다치게 하거나 사망에 이르게 하는 사건이 종종 발생한다는 것이다. 이에 여러 전문가들은 510(k) 허가 절차야 말로 ‘FDA의 가장 약하고 터무니없는 구석’이라고 비판하고 있다.
국내 많은 의료기기 업체가 FDA ‘허가’를 ‘승인’으로 고의 또는 미필적 고의로 표기하고 있어 분별해야 한다. 2등급 ‘허가’이면서 3등급 ‘승인’ 혹은 드노보 ‘승인’을 받은 것처럼 일부러 착각하게 했다면 심각한 위법 행위에 해당할 수 있다.

 목록
목록

















