
 한미약품 CHI 환우회 발표 현장사진
한미약품 CHI 환우회 발표 현장사진
한미약품은 선천성 고인슐린증 치료 혁신신약 ‘에페거글루카곤(HM15136)’의 임상 2상 경과를 발표하며, 이 치료제가 희귀질환 환자들의 삶의 질 향상에 기여할 수 있는 잠재력을 입증했다고 밝혔다.
한미약품은 최근 영국 리버풀에서 열린 유럽소아내분비학회(ESPE)에서 ‘에페거글루카곤’의 연구 성과와 임상 2상 중간 분석 결과를 공개했다. 에페거글루카곤은 선천성 고인슐린증 치료제로, 세계 최초로 주 1회 투여 제형으로 개발되고 있다.
선천성 고인슐린증은 과도한 인슐린 분비로 저혈당증을 유발하는 희귀질환으로, 현재 승인된 치료제의 한계로 인해 환자들은 부작용을 감수하거나 수술에 의존하고 있다. 한미약품은 기존 치료제의 한계를 극복할 수 있는 새로운 치료제 개발을 목표로, 에페거글루카곤의 안전성, 내약성, 약동학, 유효성을 평가하는 글로벌 임상 2상을 진행 중이다. 이 임상은 다회 용량 증량 방식으로, 공개된 데이터는 그 효과와 안전성을 뒷받침한다.
임상 2상 중간 분석 결과, 에페거글루카곤은 뛰어난 안전성과 내약성을 보였으며, 치료 중 특이한 부작용은 보고되지 않았다. 특히, 저혈당 발생 횟수와 시간이 유의미하게 감소했고, 주 1회 투약 간격을 유지할 수 있는 약리학적 근거도 확인됐다. 이러한 결과는 치료 효과의 지속성과 환자의 투약 편의성 향상에 크게 기여할 것으로 예상된다.
영국 그레이트 오몬드 스트리트 병원(GOSH)의 연구자인 안토니아 다스타마니 박사는 “에페거글루카곤의 안전성 및 유효성 프로파일이 매우 유망하다”며 "임상 참여자들의 경험을 바탕으로 이 치료제가 환자들의 삶의 질 향상에 긍정적인 영향을 미칠 것으로 기대된다"고 말했다.
 미국혈액학회에 참가해 ‘PHI-101’의 임상 1상 연구 결과를 발표한 한혜정 파로스아이바이오 미국법인 대표
미국혈액학회에 참가해 ‘PHI-101’의 임상 1상 연구 결과를 발표한 한혜정 파로스아이바이오 미국법인 대표
파로스아이바이오는 9일, 급성 골수성 백혈병(AML) 치료제 ‘PHI-101’의 임상 1상 종합 연구 결과를 발표하며, 이 치료제가 AML 환자들에게 안전성, 내약성 및 치료 효능을 보였음을 입증했다고 밝혔다.
이번 발표는 12월 7일부터 10일까지 미국 샌디에이고에서 열린 미국혈액학회(ASH)에서 이루어졌으며, 파로스아이바이는 지난해에도 ASH에서 임상 1상의 중간 결과를 발표한 바 있다. PHI-101은 기존 치료제에 내성이 생긴 재발·불응성 AML 환자에게 유망한 치료 옵션으로 평가받고 있다.
임상 1상은 서울대병원, 서울아산병원 등 다수의 대형 병원에서 30명의 재발·불응성 AML 환자를 대상으로 진행됐으며, 50%의 환자가 종합완전관해를 경험했다. 종합완전관해는 완전 관해와 불완전한 혈액학적 회복을 동반한 완전관해를 포함하는 수치로, PHI-101이 고난이도 환자들에게도 긍정적인 반응을 보였음을 보여준다. 또한, 객관적 반응률(ORR)은 67%를 기록했으며, 임상 환자들의 97%에서 FLT3 활성이 85% 이상 억제된 것으로 확인됐다.
PHI-101은 FLT3 변이를 표적해 급성 골수성 백혈병 세포의 성장을 억제하는 차세대 치료제로 개발 중이다. 파로스아이바이오는 PHI-101을 기존 치료제의 한계를 극복하는 혁신 치료제로, 2025년 글로벌 임상 2상을 시작할 예정이다. 이번 임상 시험에서 PHI-101은 FLT3 유전자 변이를 보유한 환자들에게 효과적인 결과를 보여줬으며, 이들 환자는 FLT3 변이가 없는 환자들보다 생존율이 낮고 재발 위험이 높다.
한혜정 파로스아이바이오 최고혁신책임자(CIO)는 이번 연구 결과를 통해 "PHI-101이 기존 치료제로는 효과를 볼 수 없는 환자들에게 새로운 치료 가능성을 제시했다"고 강조했다. 파로스아이바이는 PHI-101을 기존 치료제보다 뛰어난 효능을 발휘하는 차세대 급성 골수성 백혈병 치료제로 개발 중이며, 2025년 글로벌 임상 2상에 본격적으로 돌입할 예정이다.
 리가켐바이오 ASH 포스터발표내용
리가켐바이오 ASH 포스터발표내용
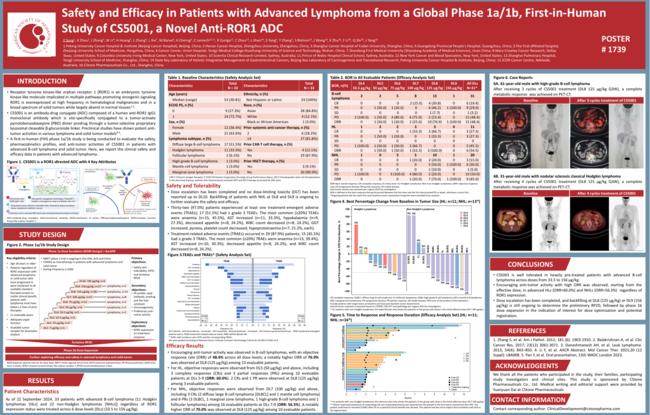
리가켐바이오의 anti-ROR1 단클론 항체 기반 ADC 치료제 CS5001이 진행성 B세포 림프종(B-cell lymphoma)과 고형암 환자들 대상으로 진행한 임상 1a/1b상에서 안전성과 항암 효능을 확인했다.
이번 연구는 CS5001이 림프종 및 고형암 모델에서 강력한 항암 효과를 보였다는 전임상 연구 결과를 바탕으로 진행됐으며, 첫 번째 임상 시험인 만큼 중요한 임상적 진전을 이뤘다. 임상 1a/1b상은 용량 증대(1a상) 및 도출된 잠정 RP2D 용량군에서의 용량 확장(1b상) 연구로 구성됐으며, 2024년 9월 22일 기준 총 33명의 환자들에게 투약이 진행됐다.
연구 결과, 용량 증대가 완료됐으며, 용량제한독성(DLT)은 보고되지 않았다. 32명의 환자들이 최소 1건 이상의 이상반응(TEAE)을 경험했으나, 주요 치료 관련 이상반응(TRAE)으로는 빈혈, 아스파트산아미노기전달효소(AST) 증가, 식욕 저하 등 경증에서 중등도 정도로 나타났다.
항암 효능 측면에서, B세포 림프종 환자들에게서 고무적인 결과가 도출됐다. 전체 용량군의 객관적 반응률(ORR)은 48.4%였으며, DL8(125μg/kg)에서 평가 가능한 13명의 환자 중 76.9%가 반응을 보였다.
호지킨 림프종(HL) 환자들의 경우, DL5(50μg/kg) 이상에서 객관적 반응이 나타났으며, DL8에서는 평가 가능한 환자들 중 100%의 ORR을 기록했다. 비호지킨 림프종(B-cell NHL) 환자들에서는 DL7(100μg/kg) 이상에서 객관적 반응이 관찰됐으며, DL8에서 70%의 ORR을 보였다.
특히, 임상 1a/1b상에서는 B세포 림프종 환자들에게서 높은 객관적 반응률(ORR)과 함께 긍정적인 항암 효과가 확인됐으며, 125μg/kg 용량군에서 78.6%의 ORR(완전 관해(CR) 4명, 부분 반응(PR) 7명)을 기록했다. 이러한 결과는 CS5001이 림프종 치료에서 유망한 후보임을 시사하며, 향후 더 넓은 적응증에 대한 임상 확장이 기대된다.
CS5001은 다수의 사전 치료 경험이 있는 환자들에서도 내약성이 우수하고 높은 항암 효능을 보여, ROR1 발현 여부와 관계없이 효과적인 항암 치료제로서의 가능성을 확인했다. 향후 이 치료제는 용량 최적화 및 허가 신청을 위한 임상 1b 용량 확대 임상에 돌입할 예정이며, 잠정 RP2D 용량을 기반으로 한 추가 연구가 기대된다.
 목록
목록





















